
Section 1.2 Evaluation
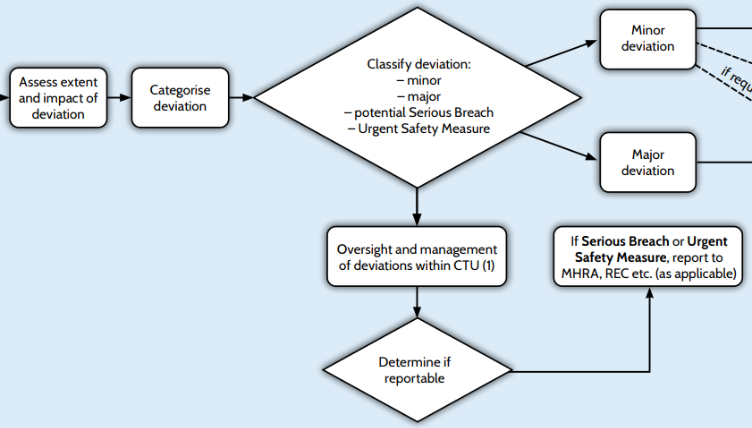
Assess Extent and Impact of Deviation

To assess a deviation, we first need to ascertain if it is a single event or multiple at one site or has occurred at several sites within a trial or across multiple trials. This, in combination with the event itself, will enable classification and action.
Standardised details may be recorded on deviation logs or trackers to assist with this assessment by specifying deviation category, and capturing information in a consistent way e.g. type of event, number of patients affected, deviation details. It is recommended practice to review the deviation log or tracker to assist with identifying whether other incidents of the same deviation type have occurred. This review will assist with classifying deviations as they occur, but also enable trend analysis to occur over time.
The trial statistician should assess the impact on data integrity and trial integrity.
Categorise Deviation

Deviations may be listed within specific categorisations such as consent, eligibility, study drug management, safety reporting, CRF completion, identifiable data, site study management, study specific procedures or processes, registration or randomisation, other CTU process and procedures. Categorisations may be predefined options within a deviation reporting form or tracker.
Classify Deviation

Standard Operating Procedures should define who has responsibility for classifying deviations. Major deviations would usually include senior staff within CTU in the decision process.
To define the path for an incident it is necessary to classify it as a protocol deviation or not and then as major or minor, including identification of events which may be (potential) serious breaches in line with your CTU SOPs.
The decision on how to classify a deviation should be predefined in SOPs or trial documentation. The classification decision will be based on a review of the deviation impact on patient safety, data integrity, GCP compliance. The classification decision may be different between trials to reflect the individual risks and impact considerations e.g. a missed protocol assessment for a Phase I trial may necessitate a different deviation classification compared to a missed protocol assessment for a Phase IV trial. It is also possible that following further review of the deviation and/or its potential impact on the trial or across trials, that the original classification may need to be amended depending on the outcome.
Classification may vary within organisations and be defined in SOPs. Example classifications are likely to include:
Minor deviations: Protocol deviation / events not affecting patient safety or data integrity, minor GCP deviation/minor non-compliance to organisational SOPs.
– Example: Patient visit out of trial window without any effect on data or trial procedures
– Example: Patient enrolled into trial by a staff member not delegated to verify eligibility (patient does meet eligibility criteria).
Major deviations: Protocol deviations with potential to affect patient safety or data integrity. It is possible that some major classifications may be considered as a potential serious breach.
– Example: Patient visit out of trial window which affects patient receiving a trial procedure.
– Example: Patient enrolled into trial but does not meet protocol eligibility criteria.
Serious Breach: Regulation 29A of the Medicines for Human Use (Clinical Trials) Regulations 2004 (Statutory Instrument 2004/1031, as amended), contains a requirement for the notification of serious breaches of GCP or the trial protocol. For the purposes of this regulation, a serious breach is a breach which is likely to effect to a significant degree – (a) the safety or physical or mental integrity of the subjects of the trial; or (b) the scientific value of the trial”.
– Example: One subject was administered 6 additional doses of IMP. The subject was to receive IMP on day 1 and 8 but instead received IMP on days 1 to 8 (The impact of SI 2006/1928).
– Example: In a CTIMP, concerns were raised during monitoring visits about changes to source data for a number of subjects in a trial, which subsequently made subjects eligible with no explanation. An audit was carried out by the Sponsor and other changes to source data were noted without explanation, potentially impacting on data integrity. Follow-up reports sent to MHRA confirmed the Sponsor concerns over consenting and data changes made to source without an adequate written explanation ( The impact of SI 2006/1928).
Non-CTIMP Serious Breach requirements are covered under the non-CTIMP Standard conditions by the NHS HRA. ‘A “serious breach” is defined as a breach of the protocol or, of the principles of Good Clinical Practice which is likely to affect to a significant degree the safety or physical or mental integrity of the research participants, or the scientific value of the research. There is no requirement to notify minor breaches of GCP or the protocol.’
Medical device: serious breaches are not defined in the medical device regulations. Note however, that all deviations to the Clinical Investigational Plan (equivalent to protocol) should be reported to the MHRA see ‘determine if reportable’ section.
Urgent Safety Measure: Regulation 30 of the Statutory Instrument (SI) 2004 Number 1031 (as amended) stipulates that the sponsor and investigator may take appropriate urgent safety measures in order to protect the subjects of a clinical trial against any immediate hazard to their health or safety.
– Example: Repeated IMP dosing incidents indicate systematic issue with instructions provided to patients – urgent safety measure (USM) to provide updated information to patients immediately. Then followed up with protocol amendment for updated documentation in line with regulatory requirements.
– Example: Ineligible patient has received protocol treatment and requires non-protocol related supportive measures.
Minor Deviation

Major Deviation

Oversight and Management of Deviations Within CTU

Deviation logs are useful to enable tracking and classification of all incidents and deviations not captured and managed elsewhere (e.g. electronic data capture tools may have in-built systems to manage data deviations). Deviation logs will be managed according to local SOPs, but may include the requirement for the deviation owner – the person who identified the deviation – to log the event. Details on the log may include deviation number, date identified (or reported to CTU), site identifier, date deviation occurred, patient ID, deviation category, deviation details, resolution details, if escalated to a CAPA, date closed.
Consideration should be given as to whether other incidents of the same deviation type have occurred either at same site, or as a trend across sites.
The trial team should review the deviation log regularly, at agreed frequency, with the review meetings/discussions being documented. For minor deviations, this review may be able to be delegated to an appropriately experienced member of the trial team. Where there are major deviations or discussion is useful, the wider CTU trial team should discuss. This may include involvement from the trial statistician(s) who will need to be kept informed and involved in decision making (e.g. closing/classifying deviations) on any events which have the potential to affect the data analysis.
Some deviations/non compliances will automatically require corrective and preventative actions (CAPA) given the seriousness of the variance from approved procedure. For example, recruiting a patient that has not met all the eligibility criteria. CAPAs should be considered when there are recurring issues or non-compliance usually at a pre-defined rate e.g. three or more occurrences of the same issue OR due to a number of individual deviations, there are concerns relating to the site management of the study.
Regardless of whether the CAPA process is used, it would usually be appropriate to consider
- Can anything be done to reduce the impact of the deviation?
- Are there lessons to be learnt at the site or at all sites or at the CTU?
- Should the protocol or site training be updated?
Determine if Reportable

The process for determining if a deviation is reportable to regulatory authorities and who should be involved in the decision-making discussions should be detailed within local SOPs as this will vary across CTUs.
For international trials, In accordance with contracted agreements, the Sponsor representative in each country that has, or will, undertake responsibility for informing the Competent Authority of any serious breaches, in that country, must be informed of all reportable deviations.
In certain circumstances, where the deviation has potentially met the criteria for escalation to a Serious Breach there are additional steps required which are not covered by this document. For further details on how to manage and report Serious Breaches, please see your Unit/Sponsors SOP for Serious Breach Reporting and also the MHRA Website: https://www.gov.uk/guidance/good-clinical-practice-for-clinical-trials#report-a-serious-breach
If Serious Breach or Urgent Safety Measure, Report to MHRA/REC, Sponsors, trial committees, and Funders (As Applicable)

Onward reporting to MHRA will be done in accordance with the relevant regulations. Urgent Safety Measures and Serious Breaches have already been referenced in the “Determine if Reportable” section.
Device trials have variation in reporting requirement compared to CTIMPs, and CTUs managing these trials should have device specific SOPs in place to account for this. There is a requirement for classification of major or minor deviations to be justified and this could be managed via the relevant SOP or trial protocol. Additionally, there is a requirement to notify the MHRA of all deviations to the study immediately, or when all deviation information is known. This can be done by the manufacturer or CTU if delegated by sponsor. Details about the nature of the deviation, when it occurred, where it occurred, and any proposed corrective actions should be provided.
Use the following MHRA protocol deviation tracker when reporting deviations and keep this as a ‘live’ document so that new deviations can be added. This enables both the sponsor and MHRA to have a complete overview each time it is submitted. The tracker can be located within the clinical investigations guidance: www.gov.uk/guidance/notify-mhra-about-a-clinical-investigation-for-a-medical-device.
It is important to ensure that onward reporting of serious breaches is detailed in relevant trial documentation, for example in the protocol as certain funders and other parties may require notification in the event of a serious breach.




